收稿日期:2017 — 08 — 16)
基金项目:1. 国家自然科学基金(81570471); 2. 天津市卫生行业重点攻关项目(14KG129)
通信作者:詹江华,zhanjianghuatj@163.com
基金项目:1. 国家自然科学基金(81570471); 2. 天津市卫生行业重点攻关项目(14KG129)
通信作者:詹江华,zhanjianghuatj@163.com
DOI: 10.3969/j.issn.1671-6353.2018.01.017
婴儿期胆汁淤积性肝病通常是由于排入十二指肠内的胆汁减少或中断引起,伴随肝内胆管进行性破坏,导致小叶间胆管数量减少或缺乏。肝内胆汁淤积主要是由于病毒感染、代谢疾病、中毒等因素引起,也可以继发于肝内胆管发育异常、肝细胞分泌胆汁功能下降。从组织病理学上,至少要看到5个汇管区,正常儿童胆管与汇管区(BD,bile duct; PT,portal tracts)比率0.9~1.8,如果低于0.5则为小叶间胆管缺乏(paucity of interlobular bile duct,PILBD)。其分为综合征和非综合征两类,前者通常伴随特殊面容、心血管系统异常、椎体发育异常; 而后者与代谢、病毒感染、染色体异常有关。目前胆道发育不良的临床诊治中还存在许多问题与疑惑,现就这一主题综述如下。
人们对于胆道发育不良的认识经历了一个漫长的过程,并且也是从认识胆道闭锁中逐步认识的胆道发育不良。胆道闭锁被认为是引起新生儿黄疸的重要原因之一。Thompson于1892年分析了50例黄疸患儿,由于当时对于胆道闭锁的诊断还很模糊,没有明确的诊断标准,究竟是胆道闭锁还是胆道发育不良,没有给出明确的定义。Izant报道美国小儿外科对843例疑似“胆道闭锁”患儿实施手术,其中12%的患儿获得正确的救治,仅6%的患儿长时间存活,这些病人是否也是胆道发育不良,没有明确的说明[1]。自此许多学者报道“胆道闭锁”患儿在未经手术治疗情况下,黄疸消退,生存时间较长。有Kanof等人于1953年将这些患儿的症状命名为浓缩胆汁综合征、胆囊狭窄或胆道部分闭锁[2]。将其命名为伴小范围肝内肝外胆道系统发育不良,这是对于胆道发育不良症最初的认识[3]。直至1964年,Longmire在国际上首次报道了35名有黄疸症状的患儿,经胆道冲洗或保守治疗后长期生存,并将该疾病正式命名为胆道发育不良[4]。
胆道发育不良根据临床表现主要分为两类,综合征型和非综合征型。
又名Alagille综合征(Alagille syndrome,AGS),累及多系统,包括肝脏、心脏、骨骼、眼睛和颜面等,是具有表型特征的慢性胆汁淤积的最常见原因。西方国家报道发病率为1/70 000~1/100 000,是一种常染色体显性遗传病,90%以上与人类染色体20p12上的JAG1基因突变有关[5],还有一些患儿存在NOTCH2基因突变[6]。
1. Alagille综合征的认识过程:由于Alagille综合征可累及身体多个脏器,各脏器表现的严重程度在不同个体之间可有很大差异,因此过去曾被称为不同的名字,包括肝动脉发育异常(arteriohepatic dysplasia,AHD)、肝管发育不全综合征(hepatic ductular hypoplasia syndrome)、综合征性小叶间胆管缺乏(syndromic paucity of interlobular bile duct)、胆汁淤积伴外周肺动脉狭窄(cholestasis with peripheral pulmonary stenosis)、Alagille-Watson Syndrome综合征(AWS)、肝内胆道闭锁(intrahepatic biliary atresia)、肝内胆管生成障碍(intrahepaffc biliary dysgenesis)等,以综合征性小叶间胆管缺乏最为常用[7]。由于以上命名均不能反映Alagille综合征的全貌,尤其部分病例甚至可不存在小叶间胆管的缺乏,因此目前Alagille综合征这一名字逐渐成为肝病、心脏和遗传学等领域中被一致认可的名字。
2. 临床表现: ①肝脏表现:肝脏上常表现为不同程度的胆汁淤积,致胆汁淤积性慢性肝病。黄疸是该病主要表现之一,多数在婴儿早期,尤其在新生儿期即可出现高结合胆红素血症,呈阻塞性黄疸表现。瘙痒是Alagille综合征的突出表现,在所有胆汁淤积性肝病中最严重,往往较黄疸和胆汁淤积表现更为明显。但可能由于感觉神经发育不成熟,患儿在3至5月龄之前很少出现此症状,幼儿期后较常见,无黄疸病人亦可有瘙痒症表现。肝肿大见于绝大部分Alagille综合征患者,包括婴儿期。脾肿大开始时少见,但随病情进展,可见于约70%的患者。因为胆汁淤积,Alagille综合征患者可有严重的高脂血症,尤其以血中胆固醇升高最明显。严重者可见多发性黄瘤,通常在生后数年内逐渐增多,随着胆汁淤积的改善可消失。肝功能检查血胆红素升高可达正常上限的30多倍,胆汁酸可达百倍以上。血中转氨酶水平也不同程度升高,但肝脏合成功能常不受影响。凝血功能障碍常见,但多在注射维生素K后纠正,表明其原因为维生素K缺乏。肝病严重程度是影响Alagille综合征病人预后的主要原因。 ②心脏表现(85%~97%):心脏杂音是该病第二常见的体征,杂音主要因肺动脉流出道或外周肺动脉狭窄引起。外周肺动脉狭窄可单独发生,也可合并心内异常,包括法洛四联症、室间隔缺损、房间隔缺损等。有报道约85%~95%的病人可见心血管异常。心血管畸形是导致Alagille综合征病人预后不良的另一主要原因。③骨骼表现(33%~87%):Alagille综合征患者可有脊椎发育异常,主要表现为蝶状椎骨。特征性的蝶状椎骨表现见于约33%~87%的患者身上。骨骼的异常通常不表现出临床症状,而在X线检查时发现。其他的骨骼异常包括指(趾)骨缩短、远端尺骨和桡骨缩短、毗连椎骨融合、第十二肋骨缺如、锥体中央透亮等。此外患儿可发生严重代谢性骨病、骨质疏松症及病理性骨折(尤其表现在股骨)等。④眼部表现(56%~88%):眼部异常涉及角膜、虹膜、视网膜及视神经乳头等。角膜后胚胎环是最具有特征性的眼部改变。角膜后胚胎环即凸出中心位的Schwalbes环,常出现在角膜内皮和色素层小梁组织的交界处。后胚胎环可见于56%~95%的患者,但8%~15%的正常人亦可见此表现,因此单独出现的诊断价值有限,只有同时存在其他异常时才有意义。其他眼部异常包括青光眼与角膜巩膜发育不全(阿克森费尔德异常)、中胚层发育不全(Rieger异常)、异常的视神经乳头、小角膜等[8]。⑤面部表现(70%~100%):Alagille综合征的面部特征为前额突出、眼球深陷伴眼距中度增宽、尖下颌、鞍形鼻并前端肥大等。特殊面容可能早在婴儿期即已存在,小婴儿以前额突出和耳发育不良多见,随年龄增长,其他各项特征逐渐突出。除了上述的5个主要表现以外,次要临床表现主要涉及肾脏、胰腺、气管或支气管、空肠、回肠和脑血管等异常。肾脏异常可见于40%~50%的Alagille综合征患者,孤立肾、异位肾、分叉型肾盂、小型肾、单侧肾、双侧多囊肾及肾发育异常等为常见表现,气管支气管狭窄、空回肠狭窄与闭锁、及小结肠等亦有报道。Alagille综合征也可有体格和精神发育障碍、大运动发育迟缓、异常的视觉、听力和其他体觉异常、肌力减退和震颤等,但多随强化营养或肝移植而改善,提示这些改变可能是继发性的。颅内出血是最重要的颅内合并症,可发生在颅内不同部位。大多数的出血发生在无显著凝血障碍的患者。头部外伤,通常是轻微的外伤也可导致颅内出血。结合最新的分子生物学研究以及尸检发现,推测其可能和固有的颅内血管发育异常有关,但这些微小的血管病变MRI也难以发现,因此目前还不能预测和预防[9,10]。良好的凝血机制纠正和头外伤后仔细观察可能降低某些病例的病死率和致残率。
1. 病因:该类型相对罕见,病因未明,推测可能是由于先天发育不良或原发胆管损伤后萎缩所致。有文献报道非综合征型小叶间胆管缺乏可由下列病因所致,包括α1-抗胰蛋白酶缺乏症、囊性纤维化、垂体机能减退、胆汁酸代谢缺陷、唐氏综合征、先天性CMV感染等[11]。具体病因情况见表1,另有一部分患儿为特发性[12,13]。
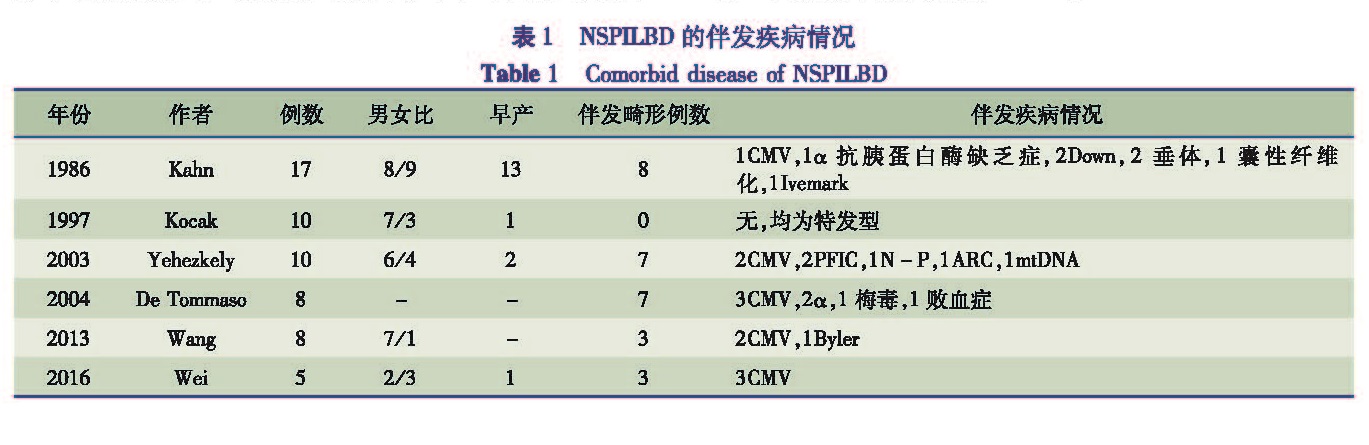
表1 NSPILBD的伴发疾病情况
Table 1 Comorbid disease of NSPILBD
2. 临床表现:主要临床表现有黄疸、无胆汁粪便。部分患儿伴有先天性发育畸形,主要是面部畸形:小颌畸形,鱼嘴,高拱嘴。还有多趾及关节弯曲变形,类人猿脚底,患儿有肾功能损害。肝脏肿大,肝酶异常,主要以丙氨酸氨基转移酶及谷氨酰转肽酶升高为主[12]。非综合征性小叶间胆管缺乏常见于患高结合性胆红素血症或JLE(青少年系统性红斑狼疮)的月龄小于1个月的患儿中,其小叶间胆管破坏开始较早(3月龄之前),且进展比综合征性胆管缺乏要快。至今没有非综合征型患儿进行基因检测的报道,因此不清楚非综合征型是否为Alagille综合征的亚临床型,还是单独的一种疾病有待进一步的研究[13]。
目前国内对于胆道发育不良的诊断和治疗报道不多,没有系统的进行诊断和治疗的描述,且从发表的文章来看,其诊治过程存在差异。
综合征型小叶间胆管缺乏经典的临床病理诊断标准:首先,胆道造影显示肝内、外胆道存在,发育较细; 肝组织活检有肝内小叶间胆管数量减少或缺如,并具有至少包括慢性胆汁淤积、心脏杂音、蝴蝶椎骨、角膜后胚胎环和特殊面容 5 个主要临床表现中的 3 个,并排除其他可能原因。如果已知有 JAG1 基因突变或家族阳性史时,2 个主要临床表现即可确诊。该诊断将病理及临床症状联合,准确率更高。AGS因其临床表现典型,故诊断可靠。有学者提出基因诊断,但由于患儿临床表现与基因型的关系多种多样,故诊断仍依赖其临床表现及组织学表现[5,7]。但基因诊断有助于产前诊断及评估活体肝脏捐献者(主要为直系亲属)符合率。
非综合征型小叶间胆管缺乏诊断标准:临床上对该类型疾病缺乏特异性诊断指标。非综合征型小叶间胆管缺乏在临床症状上可有黄疸,无胆汁便,先天性面部畸形包括小颌畸形,鱼嘴,高拱嘴。多趾,关节弯曲变形,类人猿脚底。当遇到患儿有这些症状时需高度警惕[12]。建议进一步检查。当患儿肝脏肿大,肝酶功能异常时,可做肝活检及胆道造影明确诊断。对于综合征型及非综合征型小叶间胆管缺乏,肝活检及胆道造影诊断特异性高。
胆道发育不良特点如下:①术中胆道造影有以下改变:胆囊多偏小,充盈差; 切开胆囊后见黄色胆汁(胆道尚未闭锁); 肝外胆道可显影,但纤细(胆管直径≤2 mm); 左右肝管可显影,但肝内小胆管多显影不清晰[14,15]。②术前肝活检结果:肝内小胆管减少或消失,小叶间胆管数量与汇管区的比例<0.5,对于肝内小胆管增生的可直接排除; 纤维化程度无或者轻; 肝内胆汁淤积[16]。
总之,胆道发育不良可能是一类具有共同临床特点的综合征,其诊断、治疗方法及预后均不同于胆道闭锁,对其进行早期确诊可以避免误行Kasai手术[17]。其可能的诊断依据包括: ①生后早期即出现梗阻性黄疸并持续加重; ②胆道造影:肝外胆管纤细≤2 mm,肠道有造影剂进入,肝内胆管可有细微显影但不清晰; ③肝活检:肝内淤胆表现,伴有肝内小胆管减少或消失; ④胆囊造瘘管可引流出少量胆汁[14,16]。
胆道发育不良、胆道闭锁与胆汁淤积是外科胆道畸形的三个种类。胆道发育不良:炎症累及肝外胆道,胆管上皮遭到破坏,发生纤维性变,管腔狭窄,但未完全闭塞; 胆道闭锁:胆道发育中断,胆管在某一部位盲闭,不与十二指肠相通; 胆汁淤积:胆道造影提示胆道形态正常,伴有黏稠的胆汁分泌[18]。胆道发育不良主要与胆道闭锁相似处多,故需鉴别。
胆管发育不良是否为胆道闭锁病程中的一个阶段,目前还存在较大争议。临床表现和影像学检查与胆道闭锁难以鉴别,胆总管直径<1 mm的患儿,如不予手术治疗,其预后与胆总管闭锁相似。临床过程中应认识不同疾病类型,针对病因进行治疗。
胆道发育不良常以慢性胆汁淤积为突出表现,常以出生 3个月内出现高结合胆红素血症为突出表现,跨度从轻度胆汁淤积到进行性肝衰竭,且Kasai手术不能解决其根本问题,反而加重病情。从病理表现来看,胆道发育不良常表现为肝内胆管通畅,只是数量上减少且管径变细,而不像胆道闭锁以胆管增生和胆栓形成为主。部分病例汇管区可有炎症细胞浸润,早期纤维化常不明显。若有早期纤维化,则表现为窦旁纤维化,而非汇管区纤维化,且胆道发育不良肝纤维化进程较胆道闭锁慢。但少部分的胆道发育不良病人在疾病早期可有小胆管增生,此时和胆道闭锁鉴别非常困难[20]。
对于伴有肝门部囊肿的患儿,应与囊肿型胆道闭锁进行鉴别。鉴别以下两方面内容: ①囊肿大小:这两种疾病均可以在产前做出诊断,区别是前者囊肿较大,后者囊肿相对较小。 ②胆管发育情况不同:前者是胆管已经发育,但数量减少,不伴有胆栓和肝脏纤维化; 后者是胆管上皮细胞增生,伴有胆栓和肝纤维化形成。这两种疾病术后恢复情况也有不同[21]; 有文献报道:随着年龄增长,虽然小叶间胆管的消失在大多数病例逐渐发展,但很少进展为肝硬化,与胆道闭锁最终结果不同。
目前尚无针对PILBD的病因治疗措施,一般先采用支持性及系统性内科治疗。熊去氧胆酸用于促进胆汁分泌,他汀类药物降低胆固醇水平,阿片类受体拮抗剂如利福平可帮助缓解瘙痒症状,良好的营养支持有助于改善患儿的生长发育落后情况[22]。对胆道发育不良引起的婴儿胆汁淤积症,若通过内科非手术治疗无好转,可通过胆囊造瘘、胆道冲洗方法尽早减轻肝损害、减少肝纤维化发生[23]。
Berger等[24]对1例口服熊去氧胆酸后黄疸消退效果差的6周婴儿实行腹腔镜胆囊造瘘胆道冲洗,术后第8天黄疸消退。高志刚等[25]认为,对浓缩胆栓综合征、肝炎综合征和胆道发育不良引起的阻塞性黄疸,实行腹腔镜胆囊造瘘、胆道冲洗可以减轻胆汁淤积对肝脏的损害。
虽然胆道冲洗可以解决胆汁淤积,但胆道发育不良并没有有效的手术方法,1964年,Longmir等[4]报道了离断胆总管下端后行胆囊空肠吻合的手术方法,但其效果并不明显。Kasai手术也曾被用于本病的治疗,但效果不肯定,任红霞等[26]在2004年报告了10例胆道发育不良患儿,其中7例行Kasai手术后短期疗效较好,未做进一步随访报道。Kaye等[27]对19例AGS患儿行Kasai手术并随访术后情况,得出结论Kasai手术并不能改善其预后。
有报道1例7周龄患儿通过肝活检确诊非综合征型小叶间胆管缺乏,胆道造影基本正常,后未经特殊治疗其腹水消退,胆红素、肝功能均恢复正常,在其5岁时再次行肝活检发现小胆管发育不良情况得到进一步好转[28]。因此,对于部分病情变化较慢的患儿,可长期口服复方甘草酸苷、熊去氧胆酸等药物对症治疗,定期复查。然而对于肝功能仍恶化的患儿,肝移植或许是唯一的治疗途径,也是目前公认的本病的有效疗法,可极大地改善本病的生活质量及预后。
胆管发育不良患儿经治疗后部分退黄,还有患儿可表现为黄疸加重、不退黄,或退黄而瘙痒存在。部分患儿肝脏纤维化进行性加重可出现肝硬化,甚至肝脏衰竭。对于病情严重者,肝移植是唯一的治愈方法,可改善患儿肝功能,纠正生长发育迟缓[27]。
AGS患儿活体肝移植的5年总体生存率可达80%以上。其预后仍需要移植中心及小儿外科医生进行系统规范治疗。既往研究提示,AGS患儿接受肝移植后仍有死亡发生,主要在术后30 d内,其中血管并发症、胆道并发症、肾功能衰竭和神经系统并发症被认为是术后导致患儿死亡的主要因素。因此,有学者提出AGS患儿的术前评估应包含颅内或内脏血管造影以排除颅内血管发育异常。虽然所有患儿术前均行血管造影费用昂贵,且会对患儿形成二次损伤,但临床医生在术前评估中应积极排除患儿可能存在的内脏和颅内血管畸形,避免围手术期意外的出现[10]。代谢紊乱也可以引起小叶间胆管缺乏并导致预后不良,因此,我们建议寻找一个先天性代谢缺陷的检查其中包括小叶间胆管缺乏患儿的筛查[29,30]。
综上所述,胆道发育不良疾病表现多样,最初的临床表现与胆道闭锁类似,随着年龄增长,肝脏病变加重,患儿逐渐出现胆汁淤积性肝硬化症状。AGS因具有特征性表现,诊断相对容易,NSPILBD由于其表现与胆道闭锁相似,术前明确诊断颇为困难,手术中的造影和肝脏活检对NSPILBD诊断起决定性作用。疾病诊治方法多,但预后随患儿确诊年龄的增加而越来越差,需要做肝移植的患儿其成功率也不是100%。故对于该病,我们需要做的是:做好筛查,对于疑似病例,和患儿家属做好沟通,并及时进行确诊。对于确诊患儿积极进行内外科治疗。术后用药及护理要系统规范,手术创伤对患儿亦是关键,更加需要外科大夫精湛医术及人文关怀。我们希望,未来胆道发育不良的诊断更加明确,符合循证医学的要求,治疗方法创伤更小,疾病预后会更好。
